
Can't Catch Your Breath? Calcium Channel Blockers and Pulmonary Shunting - A Deep Dive
Periodically, a patient on the unit on a nicardipine or clevidipine infusion
develops what appears to be unexplainable hypoxia. The situations are
inevitably multifactorial, but often end with a workup for pneumonia, PE, or
volume overload turning up dry and a question to me about whether it could have
been the nicardipine all along. Despite the frequency in which continuous
infusion CCBs are used in the NeuroICU this doesn't come all that often, so my
answer often boils down to "well, theoretically" - vasodilators, including
CCBs and nitroprusside1 (see also here2), have been demonstrated to
be capable of reversing hypoxia-induced pulmonary vasoconstriction, so it is
perfectly within the realm of possibility that nicardipine could contribute to
pulmonary shunting and thus new-onset hypoxia. The adverse effects are only briefly mentioned in the package inserts of either nicardipine or clevidipine, with major Phase III trials not reporting it at all. Because of the
high frequency of use and low frequency of these events, however, I have always
wondered exactly how big of a problem this really is, what patients are at the
highest risk for it, and if there is anything we can do besides turning off the
drip (which sometimes is not an option in our particularly fragile patients) and I decided to take a deep dive into the literature to see if I could find an answer.
|
Key Points
|
First, let's describe the kind of patient this happens to. I think this is best done through some illustrative case reports, of which there are six currently published in the literature.
Illustrative Case Reports
Case 1: Nimodipine
Our first case report3 comes from a familiar author
to many of us in the critical care pharmacy space and describes an unfortunate
case of a 63 year old man who was receiving nimodipine for a traumatic
subarachnoid hemorrhage after an acute decompensation which led to the
discovery of substantially expanded subarachnoid blood. After the first dose of nimodipine 60mg, the
patient's oxygen saturation declined to 68% with an accompanying decline in PaO2
from 114.0 to 32.9 mmHg. This resolved within 20 minutes after increasing FiO2
to 100% and PEEP to 7.5 mmHg, but after re-trialing the patient on nimodipine 3
days later, a similar episode occurred, with SpO2 dropping to 86% and PaO2
dropping from 89.5 to 58.7 mmHg. This also improved after increasing FiO2 to
100%.

Notably, the patient was already on 0.60 of FiO2 prior to the first episode and the patient ultimately developed ARDS the day after the second nimodipine trial, suggesting the patient had some element of pulmonary injury prior to nimodipine initiation (a pulmonary contusion was seen on the day 1 chest Xray). The patient's pulmonary status continued to decline (the worst P/F ratio reported was 91) and the patient died on day 12. The Naranjo score for nimodipine contributing to the hypoxemic events was 5 (probable) because of the repeated administration, but the degree to which nimodipine caused vs contributed to the patient's desaturations are unclear.
Case 2: Nimodipine #2
This case4 is an interesting one,
although contains little detail - an 81 year old woman is brought to the ED
hypotensive (70/45 mmHg) and hypoxemic (PaO2 42 mmHg). ED workup was largely
unremarkable, but it was revealed that the patient takes warfarin, amiodarone,
and nimodipine as an outpatient (curious choice of outpatient CCB, this is not
explored further) and there is a concern for an acute CCB overdose. After
administration of IV calcium gluconate, the patient's blood pressure rebounds
to 125/80 mmHg and PaO2 improves to 113 mmHg (with the help of FiO2 of 50%, the
delivery method of which is not explained).
While the brief case report is lacking in detail, it is an
interesting concept nonetheless - should the patient have been experiencing
shunting from her nimodipine intoxication, calcium appeared to reverse the
process.
Case 3: Nimodipine #3
The third case5 is yet another strike against
nimodipine. A 62 year old man experienced an aSAH from a ruptured AComm
aneurysm and experienced worsening hypoxia ultimately leading to intubation. On
hospital day 4, the patient's oxygenation worsens, with a PaO2 of 63 mmHg at an
FiO2 of 40%. A chest CT demonstrates posterior atelectasis out of proportion to
the P/F ratio, and a TTE demonstrated normal cardiac function without noted
anatomic shunting after an agitated saline study. The patient's PEEP was
increased to 7.5 mmHg and a follow up gas demonstrated improvement. The patient
was extubated the next day but experienced a similar worsening which led to
re-intubation which is when the relationship between desaturation and
nimodipine administration was noted.

Like the Devlin case report, this led to a high Naranjo
score (7!) and follows a similar pattern of nimodipine likely worsening some
degree of underlying pulmonary dysfunction. The pattern of desaturation was
attenuated after the nimodipine was fractionated to 30mg q2 however, suggesting
the peak effect of nimodipine may have played a role as well.
Case 4: Nicardipine
This French case report6 (lovingly translated by
Google) describes a 55 year old man admitted for C6-7 fracture that was
complicated by Pseudomonal pneumonia leading to ARDS. After the patient was
successfully treated with lung protective ventilation and underwent placement
of a tracheostomy with significant improvements in his oxygenation, he
experienced significant hypertension after waking up from sedation which was
treated with a nicardipine infusion. Soon after nicardipine initiation, the
patient experienced an acute desaturation (PaO2 111 -> 60.2 mmHg) with no
obvious cause. Nicardipine was switched to urapidil (a short acting IV alpha
antagonist used widely outside the US) and FiO2 was increased from 45% to 80% which resolved the hypoxic
event. Seven days later, a similar hypertensive episode occurred and the
patient received a single nicardipine IV 5mg dose which led to a similar hypoxic
event.
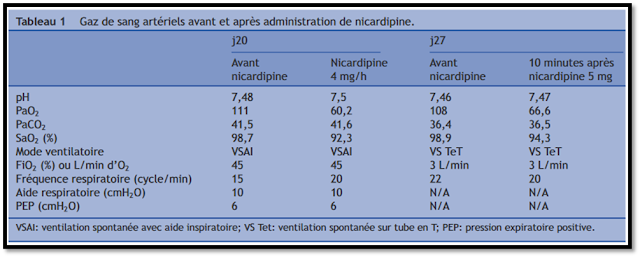
Case 5: Nicardipine #2

The second report7 involving nicardipine leaves
the neuro population and describes a 63 year old woman who underwent bilateral
orthotopic lung transplant. The patient did well post-operatively but did
experience moderate (grade 2) graft dysfunction with a P/F ratio of 215 24
hours post-op. The patient was extubated and did well on HFNC but did become
hypertensive, prompting initiation of a nicardipine infusion. The patient
experienced an acute desaturation with PaO2 dropping from 93->69 which
resolved after the discontinuation of nicardipine.
Similar to other cases, this patient had an underlying
reason to experience alveolar hypo-oxygenation of which nicardipine likely
worsened some degree of shunting. This article also has an excellent discussion
and physiology-based explanation as to why nicardipine likely contributed -
highly suggested reading!
Case 6: Clevidipine

The last illustrative case report8 involves a 16 year old boy who underwent complex AVM resection and was started on clevidipine post-operatively to reduce bleeding risk. The patient remained intubated post-operatively because a repeat surgery was planned, but 15 hours after initiation of clevidipine, the patient, who was already on an FiO2 of 60%, experienced a gradual decline in his oxygen saturation which was confirmed with a PaO2 of 33 (down from 77) 19 hours after starting clevidipine. An extensive diagnostic and therapeutic workup for the hypoxemia was negative and epoprostenol, esmolol, and paralysis with cisatracurium did not resolve the hypoxia. After consulting with their clinical pharmacist, clevidipine was identified as a potential cause, was discontinued, and the hypoxemia subsequently improved.
What is interesting about this case is that unlike the other cases, there is not an obvious underlying cause of clevidipine "tipping them over the edge", apart from likely volume resuscitation in the OR (there was only mild atelectasis on chest Xray). The dose of clevidipine here (16mg/hr) is relatively higher than the that of the nicardipine infusions used in the other cases, so there is likely an interplay between the dose and the sensitivity of the underlying substrate to shunting.
Overall, there are a number of very convincing cases that calcium channel blockers can cause clinically significant pulmonary shunting.
What is Shunting?

Shunting is likely best (or at least most comprehensively)
explained in the marvelous Radermacher9 article on gas exchange in
AJRCCM's ARDS series. In short, all blood flowing through the pulmonary
circulation either passes through ventilated lung or through non-ventilated
pulmonary tissue where the blood does not come into contact with alveolar gas.
The blood that does not participate in gas exchange is "shunted"
through the lungs, either through normal anatomic structures (bronchial and
Thebesian veins) or through physiologic structures, meaning the blood passes
through alveoli but the alveoli are either not aerated or are not capable of
participating in gas exchange. This shunted blood flow cannot be oxygenated and
will contribute to subsequent arterial hypoxemia. The total volume of pulmonary
blood flow could then be modelled as total pulmonary blood flow (Qt) is equal
to pulmonary shunt/venous admixture (Qva) plus pulmonary alveolar capillary
blood flow (Qc). In other words, the blood that gets oxygenated in the alveoli and
the blood that is shunted around the alveoli make the sum of all the blood that
flow through the lungs. The amount of blood flow made up by the shunt can be
quantified (Qva/Qt) by dividing the difference of ideal capillary O2 (estimated
at 100% in ARDS patients, lower with normal lung) and arterial oxygen
concentration by the difference in ideal capillary O2 and mixed venous oxygen
concentration. Mathematically, that would be written as Qva/Qt = (CcO2 -
CaO2)/(CcO2 - CvaO2). In practice this requires right heart catheterization to
measure pulmonary arterial blood gases but conceptually, the amount of shunt
can be thought of as how much of the "ideal" oxygen makes it to
arterial supply.

One caveat to this estimation is that shunt can either be from hypoventilated lung areas (e.g. atelectasis or pulmonary edema) or entirely nonventilated lung areas. Physiologically, this is crucial especially for the concept of whether calcium channel blockers can measurably worsen shunting - nonventilated lung areas will be entirely nonresponsive to increases in FiO2 while hypoventilated areas could have shunting partially overcome by increasing FiO2. This relationship is nicely demonstrated in the classic Benatar10 paper describing shunt lines - with significant degrees of nonventilated shunt, PaO2 will only marginally respond to increases in FiO2 because the increased oxygen delivery is simply not exposed to poorly oxygenated pulmonary blood. In this diagram, the diagonal lines represent the fraction of pulmonary blood flow made up by shunt (Qva/Qt). The X axis is the FiO2 delivered to the patient and the Y axis is the expected change in PaO2 with changing FiO2.
This becomes particularly problematic with CCBs when considering the pulmonary circulation's ability to vasoconstrict in the setting of hypoxia. Unlike the systemic circulation which *vasodilates* in the presence of hypoxemia to increase blood flow (and hopefully delivery of oxygen), the pulmonary circulation does the opposite - when pulmonary vessels are exposed to low oxygen concentrations, calcium-dependent depolarization occurs, constricting the vessel, directing blood flow *away* from the hypo-oxygenated alveoli towards better oxygenated alveoli (where vessels are not constricted). The exact mechanism of "hypoxic pulmonary vasoconstriction" (lovingly abbreviated as HPV in most articles) isn't exactly clear, with some authors11 heavily attributing the process to calcium release (both intracellular and extracellular) and some authors12 suggesting calcium plays a minor role at best. The general consensus seems to be that calcium likely plays a role, but it remains unanswered exactly how big a role and exactly how it does so.
The Physiologic Basis for CCB Shunting
While the 1980s were not the best decade for mongrel dogs,
they were remarkable for the volume of research conducted on lung physiology.
With the introduction of dihydropyridine calcium channel blockers, there was an
especially heavy focus on how these agents might improve (or worsen!) pulmonary
function in patients with pulmonary hypertension.
Long story short, the data are quite mixed on exactly what
CCBs do to pulmonary hemodynamics and oxygenation. Studies look at a few key
variables - pulmonary vascular resistance, pulmonary arterial pressure, and
arterial oxygenation. The available studies vary in the conditions in which
patients were tested, the underlying lung health, and the species the
experiments were conducted (see above re: an unfortunate decade for dogs).
Pulmonary Vascular Resistance
Most
studies evaluating the effect of calcium channel blockers objectively report
PVR values through right heart catheterization, and the overall conclusion is
that (unsurprisingly), calcium channel blockers lower PVR. This is not without
caveats, though - first, most studies (in humans) evaluate a single dose of
sublingual nifedipine. While this is generally ok to test the theory if the
drug class itself affects pulmonary circulation, it is hard to extrapolate
continuous infusions like nicardipine or clevidipine particularly because
nifedipine (especially sublingually) will increase cardiac output, affecting
the interpretation of many of these values. The second caveat is the effect on
PVR is highly dependent on the quality of the oxygen delivered to the patient
(or animal). That is, the drug response is nearly always different when the
patient is breathing room air, supplemental oxygen, or when they are hypoxic or
breathing hypoxic air (FiO2 <0.21).


In group 2, however, they compared hemodynamics in patients on room air vs those on supplemental oxygen - in patients supplemented with oxygen, nifedipine only modestly lowered PVR from baseline (from 4.2 mmHg/L/min to 3.6 mmHg/L/min) which was not significant. This suggests that when the patients' lungs were appropriately oxygenated, hypoxic pulmonary vasoconstriction was near-maximally suppressed and there was little additional room for nifedipine to vasodilate.
This general observation was confirmed in a more extreme way
Naejie et al1 - 8 healthy volunteers
underwent PA line placement and also received 20mg of sublingual nifedipine. Pulmonary
hemodynamics were then measured under two scenarios - breathing room air (FiO2
0.21) and breathing hypoxic air (FiO2 0.125). PVR was significantly lower than
baseline after nifedipine in patients breathing FiO2 0.125 air (92 to 75 dynes
per sec per cm5), but was unchanged when breathing room air (43 to 48 dynes per
sec per cm5). This has also been confirmed in animal models, with Archer et al13 finding similar results in
anesthetized dogs undergoing hypoxic challenges, Nakazawa et al14 finding similar findings in
anesthetized dogs even with cardiac output kept the same, and Johnston et al15 confirming the findings in an
experimental model of pulmonary edema in dogs. Casthely et al16 also found similar results
in anesthetized dogs undergoing intentional hypotension. The additional point
of information from these animal studies is the effect is clearly dose related,14 with higher doses of
sublingual or intravenous calcium channel blocker (with a mix of nifedipine,
nisoldipine, and nicardipine) resulting in lower PVR in the presence of
hypoxia.
Pulmonary Artery Pressure
Effects on pulmonary arterial pressure are less
well-described and appear more dependent on changes in cardiac output than the
actual vascular effect of the drug. Studies either suggest a mild increase in
PAP likely secondary to increases in CO from sublingual nifedipine (Simonneau
et al2) or no change, likely from
some equalization of vasodilation and increased CO (Naeije et al1, Melot et al17, Boldt et al18).
Arterial Oxygenation
Perhaps most importantly, nearly all studies evaluating the
effects of CCBs on pulmonary hemodynamics report the effect of the changes on
arterial oxygenation. Unfortunately, the effect is the least clear cut -
studies report decreases in PaO2, increases in PaO2, and a neutral effect on
PaO2, all depending on the drug, dose, pulmonary condition, and species(!).
Direct increases in PaO2 are the least commonly described. Boldt
et al18 report the effect of several
vasoactive medications, including both intravenous nifedipine 0.7 mcg/kg/min
and nimodipine 1 mcg/kg/min (they have all the cool drugs in Germany) in 50 men
undergoing a CABG (10 received nimodipine and 10 received nifedipine). The
authors report significant improvements in arterial oxygenation with both
nimodipine (368 to 425 mmHg) and nifedipine (382 to 424 mmHg), but it should be
noted that these patients had no underlying pulmonary dysfunction and were
ventilated with aggressive oxygenation (FiO2 of 1.0 and PEEP of 5 mmHg
throughout the procedure), so changes in PaO2 likely reflect an oxygen rich
environment supported by a mild increase in cardiac output from the CCBs and
these findings would be difficult to generalize to an ICU population.
Neutral effects on PaO2 are also relatively uncommon in the
experimental realm - Naeije et al1 did not observe lower PaO2 in
healthy volunteers breathing FiO2 of 0.125 who received nifedipine vs control
(41 vs 41 mmHg), but these patients had no underlying pulmonary pathology and
intact compensatory mechanisms - the lower PVR was compensated by increased
cardiac output. Nakazawa et al14 also reported a neutral
effect of nicardipine on arterial oxygen in their population of anesthetized
dogs, even when cardiac output was kept constant. This is in the context of
only the left lower lobe of the lung being exposed to nicardipine, however, so
sufficient lung area may not have been involved to affect systemic oxygenation.

A potential decrease in PaO2 appears to be more expected
especially in the setting of underlying respiratory dysfunction. Worsening
oxygenation was noted in Simonneau et al2 in patients with COPD not
receiving oxygen (42 to 38 mmHg, p < 0.05) but not when supplemental oxygen
was delivered (227 to 224 mmHg, p >0.05) and oxygenation worsened in a
dose-related fashion in Archer et al's13 group of anesthetized dogs
with nisoldipine specifically, but only when the dogs were intentionally made
hypoxic (36 mmHg at baseline to 35 with low dose, 32 with medium dose, and 28
mmHg with high dose nisoldipine). Casthely et al16 and Johnston et al15 also noted worsening
oxygenation in their animal models, but interesting Johnston et al, which used
a oleic acid model of pulmonary edema, noted that while PaO2 was lower (from a
baseline of 177 torr to 67 torr, although this was up form when the initial
oleic acid injury occurred), total *delivery* of oxygen increased, likely
secondary to improvements in CO.
Overall, the effect here appears to be more complex are hard
to predict. In patients who are already hypoxic, it appears a calcium channel
blocker has the potential to worsen the underlying hypoxia. In patients with
pristine lung function without risk factors for hypoxia, the evidence suggests
that a CCB is going to induce de novo hypoxemia.
Shunt
To round out the discussion, it's important to recognize
whether new hypoxia observed in patients on a CCB is directly attributable to
an actual worsening of right to left shunt. Quantifying shunt is challenging,
however, and thus few studies actually report what percent of pulmonary blood
flow is made up of shunt. In patients with underlying *pathologic* shunt, it
does appear that CCBs have the potential to improve right to left shunt - Melot
et al17 report on two patients with
underlying PAH with one patient who had an average V/Q ratio of 2.6 (ideal is
~0.8) and 20% of blood flow being made up of shunt. While sublingual nifedipine
did shift more lung units towards worsening V/Q mismatch (18.9% of lung units
had a V/Q ratio <0.2 vs a baseline of 11.9%), the overall distribution
shifted closer to an ideal ratio, with overall shunt decreasing to 10%. Boldt
et al18 reported no effect of
nimodipine or nifedipine on shunt percent (Qs/Qt of 15% at baseline to 16.2%
with nimodipine and 15.7% to 17.9% with nifedipine, p>0.05), but as noted
above, these patients were hyperoxygenated which may have attenuated the
ability to detect a change.
Quantification of worsening shunt was observed in two of the
animal model experiments. Both Casthely et al16 and Nakazawa et al14 reported significant
increases in total pulmonary blood flow being made up by shunt, with Casthely
revealing a doubling (9.7% -> 20.5%) after administration of nifedipine to
dogs and Nakazawa similarly reporting a doubling (10% -> 20.4%) with
nicardipine, but notably only in dogs who were made intentionally hypoxic and
with constant cardiac output. At a fixed cardiac output, shunt did increase,
but only mildly (9.7% -> 13.1%). Additionally, when kept at a constant
cardiac output, the effect was also dose dependent, with higher doses (6
mcg/kg/min) leading to a larger shunt percentage than lower doses (1 and 3
mcg/kg/min).
While data are more limited on the quantification of shunt
percentage, it stands to reason that this acts similarly to what was found with
arterial oxygenation. In patients with derecruited lung units contributing to
hypoxia, a calcium channel blocker likely will worsen underlying shunt which is
what is driving worsening hypoxia. In patients with fully recruited lungs,
however, a calcium channel blocker is unlikely to worsen what little shunt
already exists.
What to Do if it Happens
There are a few insights from the available data which can help to develop an action plan for an acutely hypoxic patient on a calcium channel blocker. I listed a few of the things I gleaned from my reading which will hopefully help you if you're faced with a similar situation:
- Calcium channel blockers are unlikely to cause hypoxia on their own with absolutely no inciting cause. Make sure you do your due dilligence evaluating the standard causes of hypoxia before chalking the hypoxia up to the calcium channel blocker, because even if stopping the nicardipine or clevidipine improves things, there may still be something underlying that you may have missed.
- Reasonable lung recruitment maneuvers may be enough to reverse the hypoxia. If shunt is being caused by atelectasis, recruiting additional lung units may be enough to abolish the hypoxic pulmonary vasoconstriction being inhibited by the calcium channel blocker and reverse the process.
- The effect is dose related, but there isn't clear evidence that the effect is larger with one calcium channel blocker vs another (apart from one study that suggested it was particularly notable with nisoldipine, but who uses that?). If your patient needs a calcium channel blocker (i.e. nimodipine in aSAH), try lower doses or fractionated doses so the patient sees a lower peak concentration. For continuous infusions, there may be a continuous infusion rate that achieves SBP goals but doesn't affect PVR, especially if combined with other agents. Also, because of the relation to peak effect, amlodipine is unlikely to be an issue here.
- While not discussed at length here, this effect is not universal to all blood pressure agents. While it is also observed with nitroprusside (which should essentially never be used in the NeuroICU), labetalol does not affect that pulmonary circulation to a significant degree and is a potential alternative continuous infusion if a CCB can't be used.
- You can absolutely rechallenge someone with a CCB if they experienced hypoxia previously. Because the effects depend on the underlying state of the lungs, if someone is otherwise improved (say extubated after their prior event with a CCB) and needs tight blood pressure control, you could trial a CCB again.
Conclusion
Calcium channel blockers can cause shunting and hypoxia, but
it's more complicated than it may seem at face value. While calcium channel
blockers are active in the pulmonary vasculature and can inhibit hypoxic
vasoconstriction which can worsen shunting, they will not cause substantial
shunting and hypoxia without underlying vasoconstriction already happening.

Clinical Pharmacist, Neurocritical Care
Massachusetts General Hospital
Ajwebb@mgh.harvard.edu
References
1. Naeije R, Mélot C,
Mols P, Hallemans R. Effects of vasodilators on hypoxic pulmonary
vasoconstriction in normal man. Chest. 1982;82(4):404-410.
doi:10.1378/chest.82.4.404
2. Simonneau
G, Escourrou P, Duroux P, Lockhart A. Inhibition of Hypoxic Pulmonary
Vasoconstriction by Nifedipine. New England Journal of Medicine.
1981;304(26):1582-1585. doi:10.1056/NEJM198106253042606
3. Devlin
JW, Coplin WM, Murry KR, Rengachary SS, Wilson RF. Nimodipine-induced acute
hypoxemia: case report. Neurosurgery. 2000;47(5):1243-1246; discussion
1246-1247. doi:10.1097/00006123-200011000-00048
4. Gerloni
R, Copetti R. Easily reversible hypoxemia and hypotension induced by
nimodipine. Eur J Emerg Med. 2004;11(5):295-297.
doi:10.1097/00063110-200410000-00012
5. Baker
M, Bastin MT, Cook AM, Fraser J, Hessel E. Hypoxemia associated with nimodipine
in a patient with an aneurysmal subarachnoid hemorrhage. Am J Health Syst
Pharm. 2015;72(1):39-43. doi:10.2146/ajhp140196
6. Cotte
J, D’Aranda E, Esnault P, Bordes J, Meaudre E. Hypoxie sous nicardipine :
rôle de la vasoconstriction pulmonaire hypoxique. Revue de Pneumologie
Clinique. 2012;68(3):221-224. doi:10.1016/j.pneumo.2011.08.003
7. Mishra
A, Reed RM, Eberlein M. Severe, Rapidly Reversible Hypoxemia in the Early
Period after Bilateral Lung Transplantation. Ann Am Thorac Soc.
2016;13(6):979-985. doi:10.1513/AnnalsATS.201602-107CC
8. Short
JH, Fatemi P, Ruoss S, Angelotti T. Clevidipine-Induced Extreme Hypoxemia in a
Neurosurgical Patient: A Case Report. A A Pract. 2020;14(2):60-62.
doi:10.1213/XAA.0000000000001146
9. Radermacher
P, Maggiore SM, Mercat A. Fifty Years of Research in ARDS.Gas Exchange in Acute
Respiratory Distress Syndrome. Am J Respir Crit Care Med.
2017;196(8):964-984. doi:10.1164/rccm.201610-2156SO
10. Benatar
SR, Hewlett AM, Nunn JF. THE USE OF ISO-SHUNT LINES FOR CONTROL OF OXYGEN
THERAPY. British Journal of Anaesthesia. 1973;45(7):711-718.
doi:10.1093/bja/45.7.711
11. Weigand
L, Foxson J, Wang J, Shimoda LA, Sylvester JT. Inhibition of hypoxic pulmonary
vasoconstriction by antagonists of store-operated Ca2+ and nonselective cation
channels. Am J Physiol Lung Cell Mol Physiol. 2005;289(1):L5-L13.
doi:10.1152/ajplung.00044.2005
12. Robertson
TP, Hague D, Aaronson PI, Ward JPT. Voltage-independent calcium entry in
hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J
Physiol. 2000;525(Pt 3):669-680. doi:10.1111/j.1469-7793.2000.t01-1-00669.x
13. Archer
SL, Yankovich RD, Chesler E, Weir EK. Comparative effects of nisoldipine,
nifedipine and bepridil on experimental pulmonary hypertension. J Pharmacol
Exp Ther. 1985;233(1):12-17. PMID: 3872360
14. Nakazawa
K, Amaha K. Effect of nicardipine hydrochloride on regional hypoxic pulmonary
vasoconstriction. Br J Anaesth. 1988;60(5):547-554.
doi:10.1093/bja/60.5.547
15. Johnston
WE, Vinten-Johansen J, Tommasi E. Effect of nifedipine on oxygen delivery in
canine asymmetric oleic acid lung injury. Crit Care Med.
1990;18(7):738-743. doi:10.1097/00003246-199007000-00012
16. Casthely
PA, Villanueva R, Rabinowitz L, Gandhi P, Litwak B, Fyman PN. Intrapulmonary
shunting during deliberate hypotension with nifedipine, diltiazem and labetalol
in dogs. Can Anaesth Soc J. 1985;32(2):119-123. doi:10.1007/BF03010034
17. Mélot
C, Naeije R, Mols P, Vandenbossche JL, Denolin H. Effects of nifedipine on
ventilation/perfusion matching in primary pulmonary hypertension. Chest.
1983;83(2):203-207. doi:10.1378/chest.83.2.203
18. Boldt
J, Von Bormann B, Kling D, Ratthey K, Hempelmann G. Influence of nimodipine and
nifedipine on intrapulmonary shunting--a comparison to other vasoactive drugs. Intensive
Care Med. 1987;13(1):52-56. doi:10.1007/BF00263558


Comments
Post a Comment